Introduction
In 2006, Takahashi and Yamanaka made the groundbreaking discovery that ectopic expression of four factors, Oct4 (octamer-binding transcription factor 4), Sox2 (SRY-box2), Klf4 (Kruppel-like factor 4) and c-Myc, could reprogram terminally differentiated cells into induced pluripotent stem cells (iPSCs). These cells have infinite expansion potential and share genetic markers, epigenetics and multilineage differentiation capacity with embryonic stem cells (ESCs)¹.
Unlike ESC research, which requires human blastocysts, iPSCs can be generated from an adult skin biopsy, eliminating major ethical concerns. iPSCs are now widely used in both research and clinical applications.
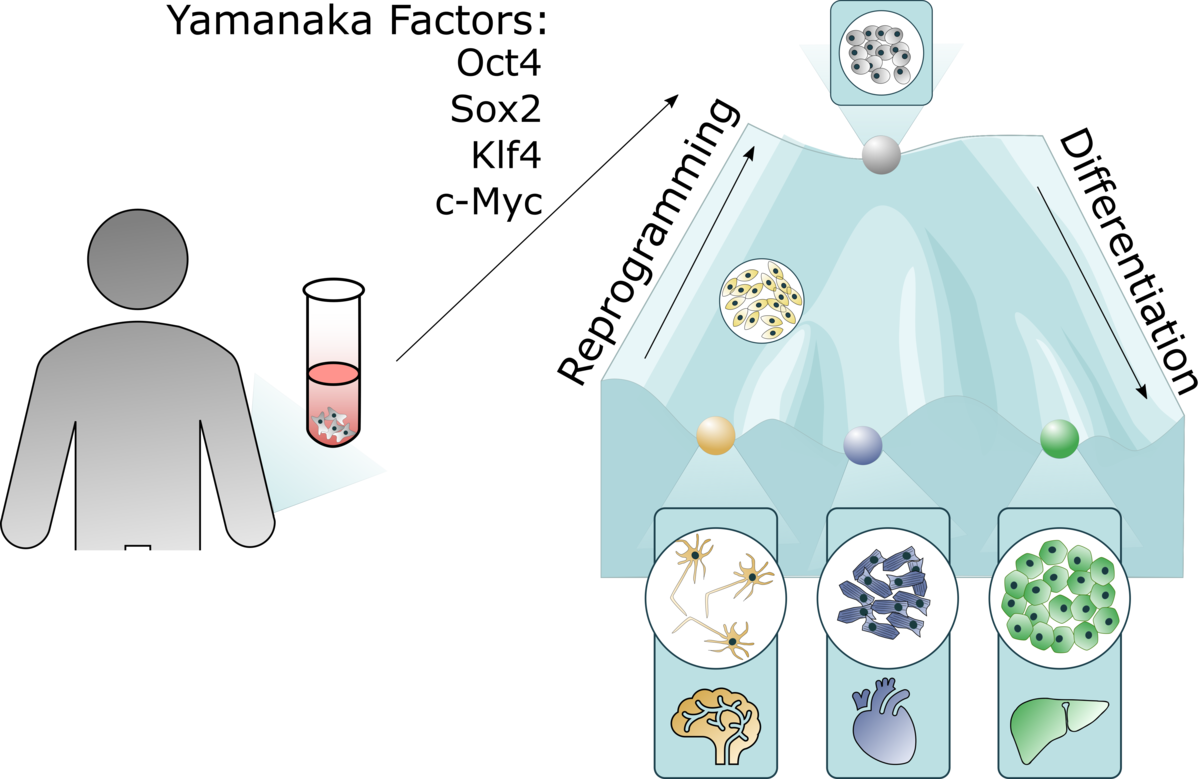
iPSCs as research models
The ability to revert differentiated cells to a stem cell state, and then guide them into specific lineages, has revolutionised biomedical research. iPSCs are powerful tools for disease modelling, drug discovery and toxicology studies.
Disease modelling
Previously, disease modelling relied heavily on overexpression or knockout approaches in immortalised cell lines, often unrelated to the tissue of interest, combined with post-mortem tissue analysis. These methods provided limited insight, as they failed to capture disease progression at early stages. Animal models offered closer physiology, but significant limitations remained, alongside growing concerns about animal welfare.
With iPSCs, patient-derived cells can be reprogrammed to mimic diseases in vitro. For example, iPSC-derived cardiomyocytes from a patient with hypoplastic left heart syndrome (HLHS) revealed defects in differentiation, sarcomere organisation and contractility that explained the clinical phenotype².
Neuroscience has particularly benefited from iPSC-based “disease-in-a-dish” models. Disorders such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis (ALS), Huntington’s disease, Machado–Joseph disease and schizophrenia can now be studied in patient-derived neural cells. iPSCs faithfully reproduce hallmarks such as α-synuclein accumulation, while also uncovering novel dysfunctions like impaired axonal transport and neurite extension³.
These models, coupled with advances in genome sequencing and engineering, have accelerated research into rare diseases. Mutations can be identified and directly linked to phenotypes using CRISPR/Cas9 to introduce or correct variants in iPSC lines. Retinal research has benefited greatly, with complex models such as retinal pigment epithelium (RPE) and even mini-retinas enabling studies of tissue development, ageing and disease pathways.
Drug discovery and toxicology
iPSCs also play a central role in drug development. They provide relevant, scalable cell models suitable for high-throughput screening (HTS) with readouts such as luciferase or GFP. For instance, ALS patient-derived motor neurons were used to screen compound libraries, identifying bosutinib, a chronic myelogenous leukaemia drug, as protective against neuronal death³.
iPSC-derived models also allow researchers to reassess existing therapies. In schizophrenia, patient-derived neurons revealed reduced PSD95 and glutamate receptor expression. Testing five common antipsychotics showed that only one improved neural connectivity.
Toxicology studies have similarly advanced. iPSC-derived cell types mimic human tissues more accurately, improving prediction of toxicity profiles, reducing reliance on animal studies, and helping avoid costly late-stage trial failures.
iPSCs as therapeutic tools
Initial therapeutic approaches focused on autologous therapies: reprogramming a patient’s own cells, correcting mutations, differentiating them into the required lineage, and reinfusing them. However, the timeframes involved are impractical for many diseases.
Attention is shifting toward allogeneic “off-the-shelf” therapies. Challenges include immune rejection due to HLA mismatch. Strategies to address this include creating large iPSC banks, such as Japan’s project aiming for coverage of 95% of the population, which requires more than 140 lines. Another strategy is the development of universal donor lines lacking B2M and CIITA, thereby eliminating HLA class I and II expression.
Clinical trials using iPSC-derived cells are underway in the US, China, Germany and Japan, targeting conditions ranging from AMD and Parkinson’s disease to spinal cord injuries and aplastic anaemia¹³.
Table 1. Examples of diseases targeted in iPSC-based clinical trials.
| Disease | Cell type |
|---|---|
| AMD (Age-related macular degeneration) | Retinal pigment epithelium (RPE) |
| RP (Retinitis pigmentosa) | Retinal sheet |
| Limbal stem cell deficiency | Limbal epithelial stem cells |
| Parkinson’s disease | Dopamine neural progenitor cells |
| Spinal cord injuries | Neural progenitor cells |
| Heart failure | Cardiomyocytes |
| Aplastic anaemia | Platelets |
| Thrombocytopenia | Megakaryocytes |
| Advanced head & neck cancer | iNKT cells |
| Articular cartilage damage | Chondrocytes |
| Urea cycle disorder | Hepatocytes |
| Chronic kidney disease | Nephron progenitor cells |
| Recessive dystrophic epidermolysis bullosa | Keratinocytes |
| Multiple sclerosis | Oligodendrocyte precursor cells |
Despite their potential, iPSCs present challenges. They may retain epigenetic memory, influencing differentiation⁴. Stringent quality control is essential to exclude residual pluripotent cells, which pose a tumourigenic risk. Additionally, assessing cell maturity is critical, as immature cells can cause adverse outcomes, such as arrhythmias from immature cardiomyocytes.
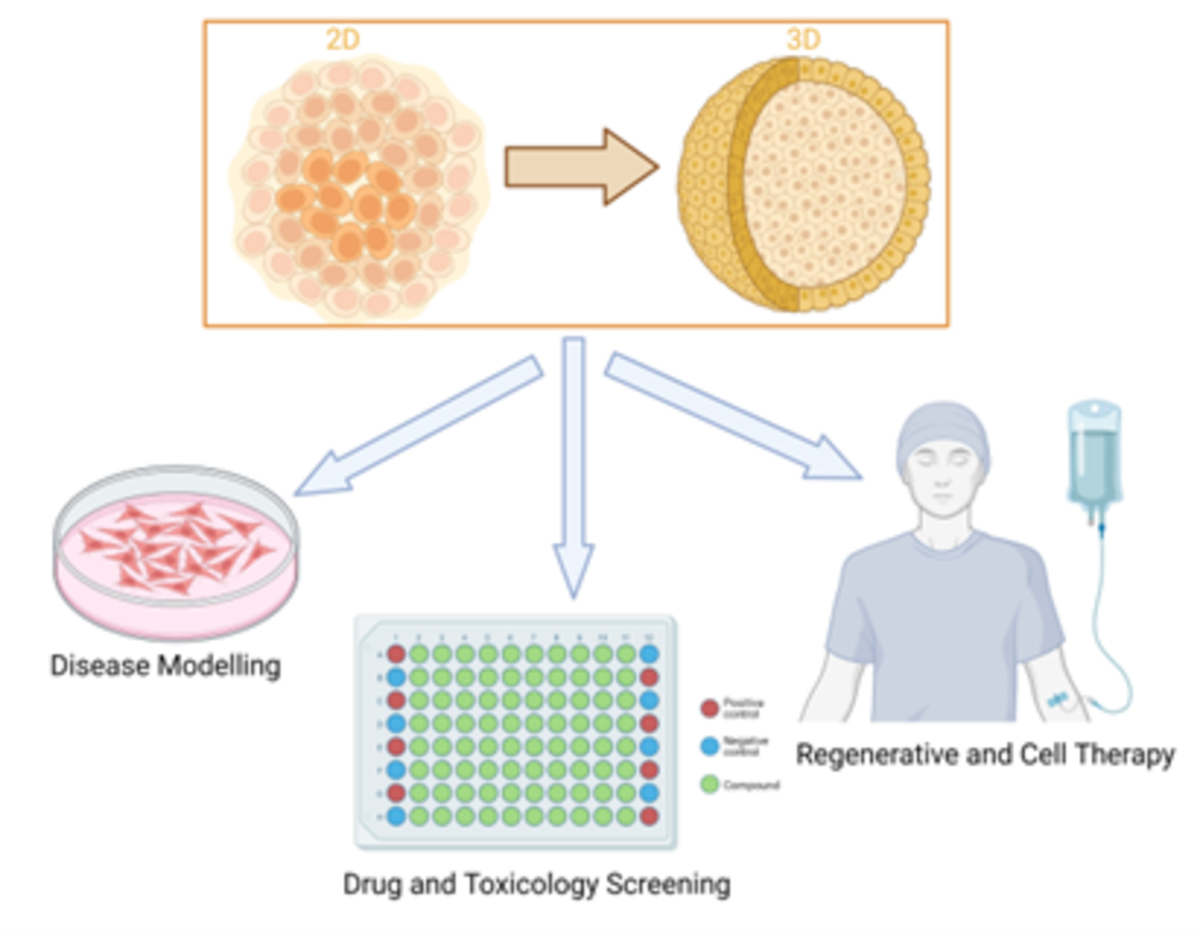
The advent of iPSC-derived organoids
Traditional 2D cultures and spheroids fail to recapitulate tissue complexity. iPSCs, however, can self-organise into organoids, three-dimensional structures mimicking tissue architecture, heterogeneity and interactions. Organoids have been developed for retina, brain, intestine, liver and kidney⁶.
Applications vary. Developmental studies aim to mimic in vivo conditions. Regenerative medicine focuses on scalability. Drug screening requires recapitulating key aspects of tissue biology with simple readouts⁵.
Reporter systems such as luciferase enhance screening, while technologies like microencapsulation and organ-on-chip platforms enable HTS. Organoids show high predictive value for drug responses, representing a major step forward in translational research⁷.
Conclusions
iPSCs continue to transform both research and therapy. Future advances in modelling late-onset diseases, vascularisation, and spatial–temporal differentiation control will improve therapeutic outcomes.
BPS Bioscience provides iPSC lines with reporter genes such as GFP and luciferase for easy readouts in HTS screening and toxicity studies, as well as Cas9-expressing iPSCs for efficient genome editing, making them ideal tools for disease modelling and therapeutic development.
References
Okano, H. & Yamanaka, S. Induced pluripotent stem cells: past, present, and future. Mol. Brain7, 22 (2014).
Grskovic, M. et al. Induced pluripotent stem cells — opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov.10, 915–929 (2011).
Tsujimoto, H. & Osafune, K. Recent progress in the applications of iPS cell technology for disease modelling and drug discovery. FEBS J.289, 7274–7291 (2022).
Yuasa, S. et al. Retained epigenetic memory in iPS cells: implications for differentiation. Front. Cell Dev. Biol.9, 831304 (2022). doi:10.3389/fcell.2021.831304
Yin, X. et al. Engineering organoids to model human development and disease. Cell Stem Cell18, 25–38 (2016).
Ye, L. et al. iPSC-derived cardiomyocytes and organoids: new frontiers in cardiac research. Curr. Cardiol. Rev.9, 63–72 (2013).
Kruczek, K. et al. Retinal organoids from human pluripotent stem cells: functional models for development and therapy. Stem Cell Reports16, 252–263 (2021).
Supplier
BPS Bioscience
Experts in protein design, expression, purification, and characterization, cell line and lentivirus engineering, and biochemical and cellular assay development