Wastewater-Based Epidemiology (WBE) uses wastewater to analyze community spread, as it is known that SARS-CoV-2 is shed through bodily excretions. Since both symptomatic and asymptomatic individuals contribute to wastewater inputs, we hypothesized that the resultant pooled sample of population-wide excreta can provide a more comprehensive picture of SARS-CoV-2 genomic diversity circulating in a community than clinical testing and sequencing alone.
Using the Swift Normalase Amplicon SARS CoV-2 Panel (SNAP) for variant analysis
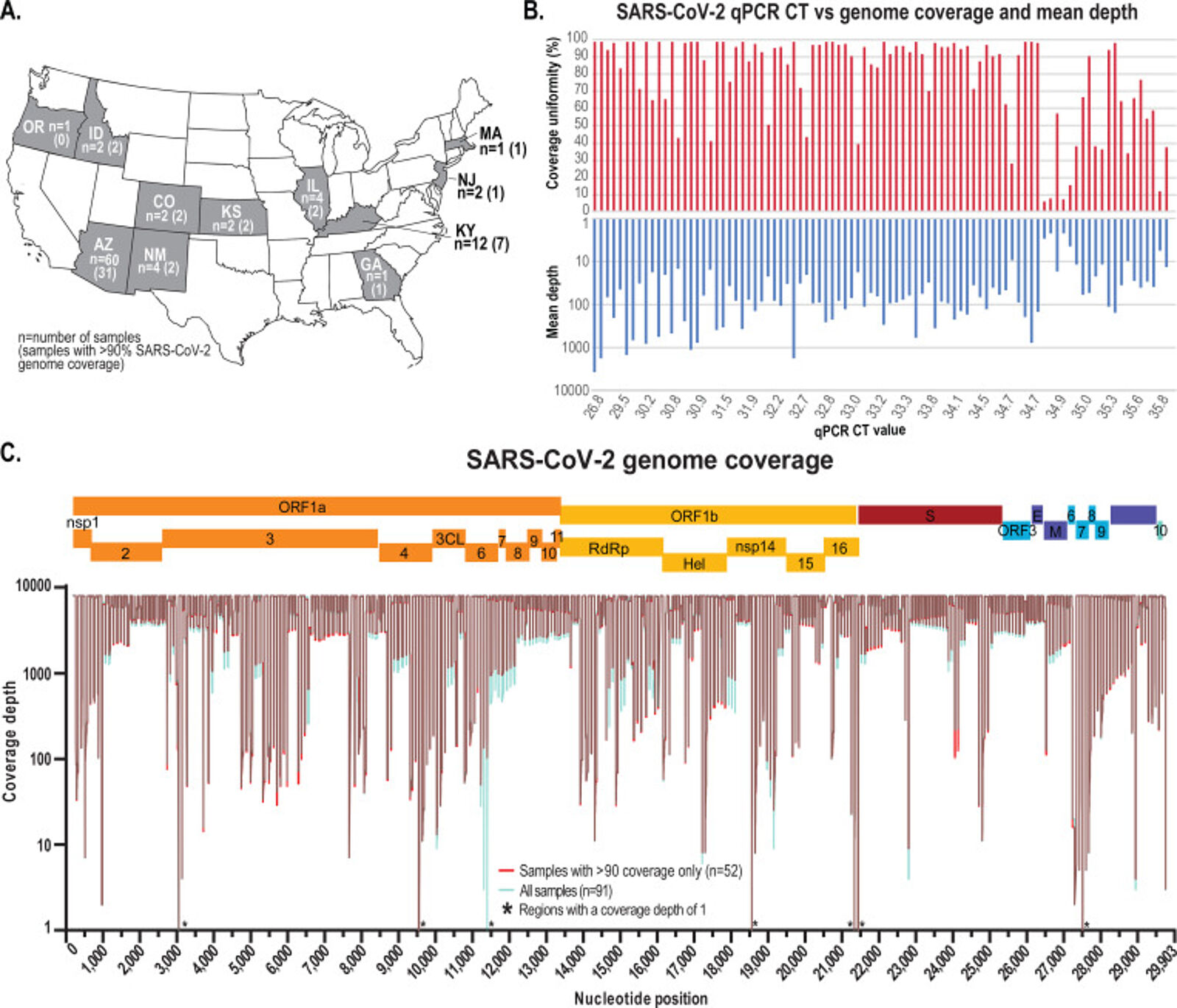
91 positive samples from 11 states in the USA were collected, plus one negative control sample. The samples were processed for high-throughput SARS-CoV-2 amplicon sequencing using Swift'sAmplicon SARS CoV-2 Panel (SNAP) on an Illumina® HiSeq 2500 sequencer.
The wastewater-derived SARS-CoV-2 sequence data indicates there were more lineages circulating across the sampled communities than represented in the clinical-derived data. Principal coordinate analyses identified patterns in population structure based on genetic variation within the sequenced samples, with clear trends associated with increased diversity likely due to a higher number of infected individuals relative to the sampling dates. The authors demonstrated that genetic correlation analysis combined with SNVs analysis using wastewater sampling can provide a comprehensive snapshot of the SARS-CoV-2 genetic population structure circulating within a community, which might not be observed if relying solely on clinical cases.