In a breakthrough effort, scientists at the Children’s Hospital of Philadelphia, Aldevron and Integrated DNA Technologies have developed the world's first personalized CRISPR-based gene therapy in just six months. The therapy was developed to treat an infant patient with severe carbamoyl-phosphate synthetase 1 deficiency, a hereditary disease with a greater than 50% mortality rate in infants.
What is carbamoyl-phosphate synthetase 1 (CPS1) deficiency?
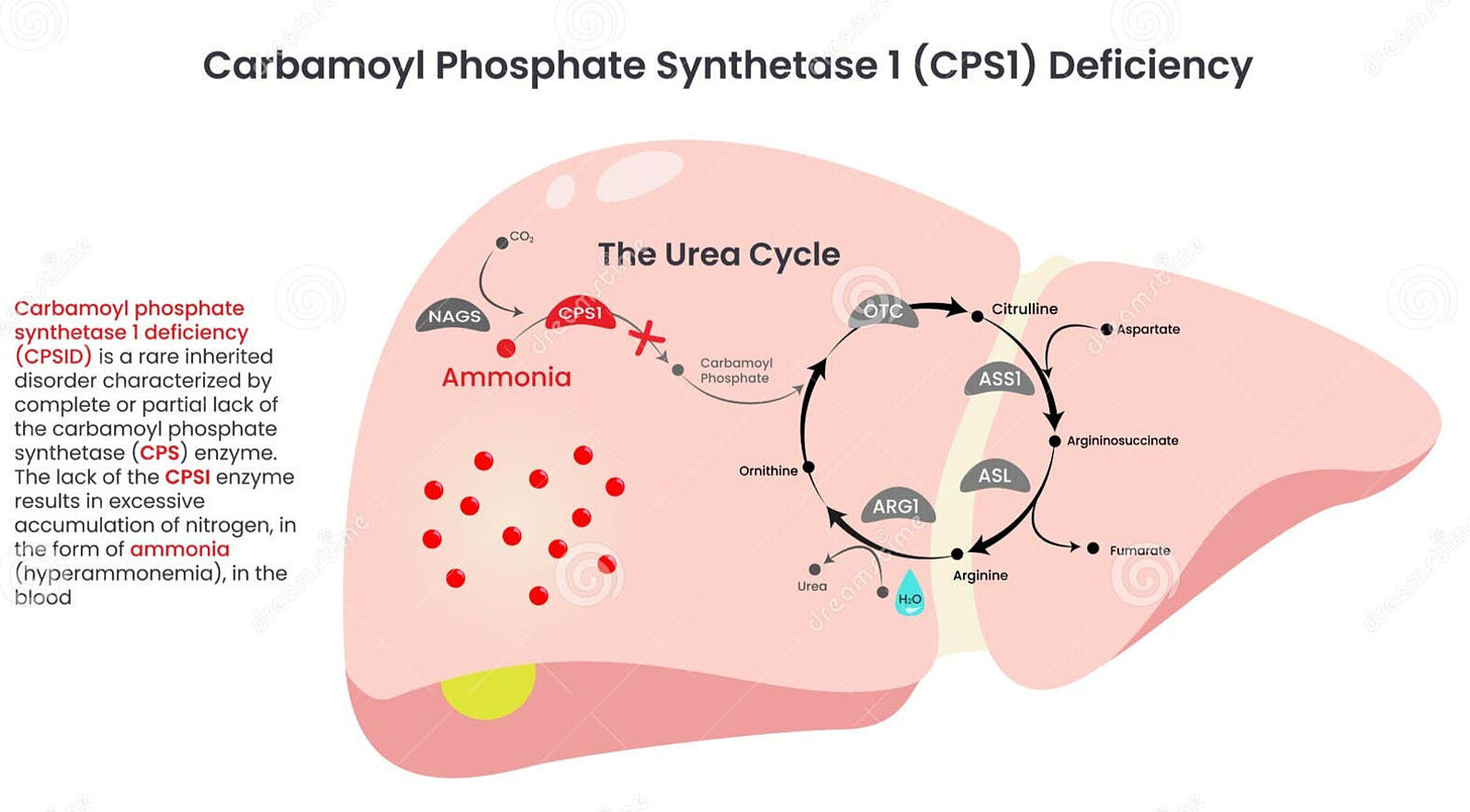
Carbamoyl-phosphate synthetase 1 (CPS1) deficiency is a rare inherited metabolic disorder that affects the urea cycle, a critical biochemical pathway responsible for removing ammonia (a toxic byproduct of protein metabolism) from the body. It's cause are mutations in the CPS1 gene, which encodes the enzyme carbamoyl-phosphate synthetase 1. This enzyme is essential for the first step of the urea cycle, where it converts ammonia + bicarbonate into carbamoyl phosphate. Without functional CPS1, ammonia accumulates in the blood (hyperammonemia), leading to brain damage, coma and death if untreated. There is currently no cure for CPS1 deficiency and treatment focuses on managing ammonia levels in the blood using scavengers such as sodium benzoate or sodium phenylacetate, hemodialysis, low protein diet and ultimately, liver transplantation.
The treatment
After the neonate patient had received a diagnosis of severe carbamoyl-phosphate synthetase 1 deficiency, scientists at Aldevron and Integrated DNA Technologies immediately began to develop a customized lipid nanoparticle–delivered base-editing therapy in order to correct the defect in the CPS1 gene. The technically complex therapy required a new guide RNA (gRNA) sequence, a new mRNA-encoded base editor, custom off-target safety services and a clinically validated lipid nanoparticle (LNP) formulation.
After regulatory approval had been obtained for the therapy, the patient received two infusions at approximately 7 and 8 months of age. 7 weeks post infusion, the patient was able to receive an increased amount of dietary protein and a reduced dose of a nitrogen-scavenger medication, without major adverse events. The patient will now be monitored in a long-term follow-up to assess safety and efficacy of the new treatment.
“This study is an important milestone,” said corresponding author Kiran Musunuru. “The impact of this work extends beyond this particular patient and category of clinical indications—it suggests a potential roadmap for transforming CRISPR therapies for other inborn errors of metabolism and life-threatening genetic diseases. It’s an exciting future for personalized medicine.”
“What we’ve accomplished together sets a new gold standard for operationalizing the future of medicine,” said Sandy Ottensmann, VP/GM, Gene Writing & Editing at IDT. “The implications of this work are profound and illuminate how collaborations between academic medicine and industry can enable major science wins. The opportunity ahead lies in continuously leveraging our CRISPR toolbox to innovate and operationalize more potential treatments and help patients in desperate search of cures.”
References
Musunuru et al. (2025) NJEM. DOI: 10.1056/NEJMoa2504747