The group of Prof. Paola Picotti at ETH Zurich has developed a mass spec-based approach for detecting protein structural and functional changes at proteome scale.
Published last month in Cell, the approach uses limited proteolysis-mass spectrometry (LiP-MS) to investigate structural and functional changes across large numbers of proteins and with high spatial resolution, offering another dimension of protein data beyond the expression-based measurements that have traditionally dominated proteomics research.
LiP-MS combines digestion with a broadly specific protease followed by a standard mass spec-based proteomics workflow to assess structural changes on a proteome-wide scale. The basic notion underlying the approach is that in the initial digestion step, the protease will cleave proteins only at sites that are accessible, left exposed by whatever structural conformation it happens to be in at the time of analysis. When a sample is treated with an agent like a drug or otherwise altered, the proteins that bind to this molecule will undergo a structural change, and this will be reflected in changes where the protease is able to cleave the protein.
By following this initial digestion step with trypsin digestion and mass spectrometry analysis, they were able to compare the peptides generated in treated and untreated samples and, based on changes in the peptides produced, determine which proteins had their structures altered by the treatment in question.
They used the technique to simulteneously look at protein structural changes during different cellular events and at the functional implications of those changes. Because LiP-MS allows for parallel measurement of protein expression and structural changes, the researchers were thus able to assess the contributions of both, capturing a broad range of functional alterations.
Subsequently, Picotti's team looked at the response in yeast to heat and osmotic stress. They found that protein structural changes were far more abundant than expression changes. For instance, only around 1 percent of detected proteins in yeast showed abundance changes, whereas 23 percent and 11 percent showed structural changes in response to heat shock and osmotic shock, respectively.
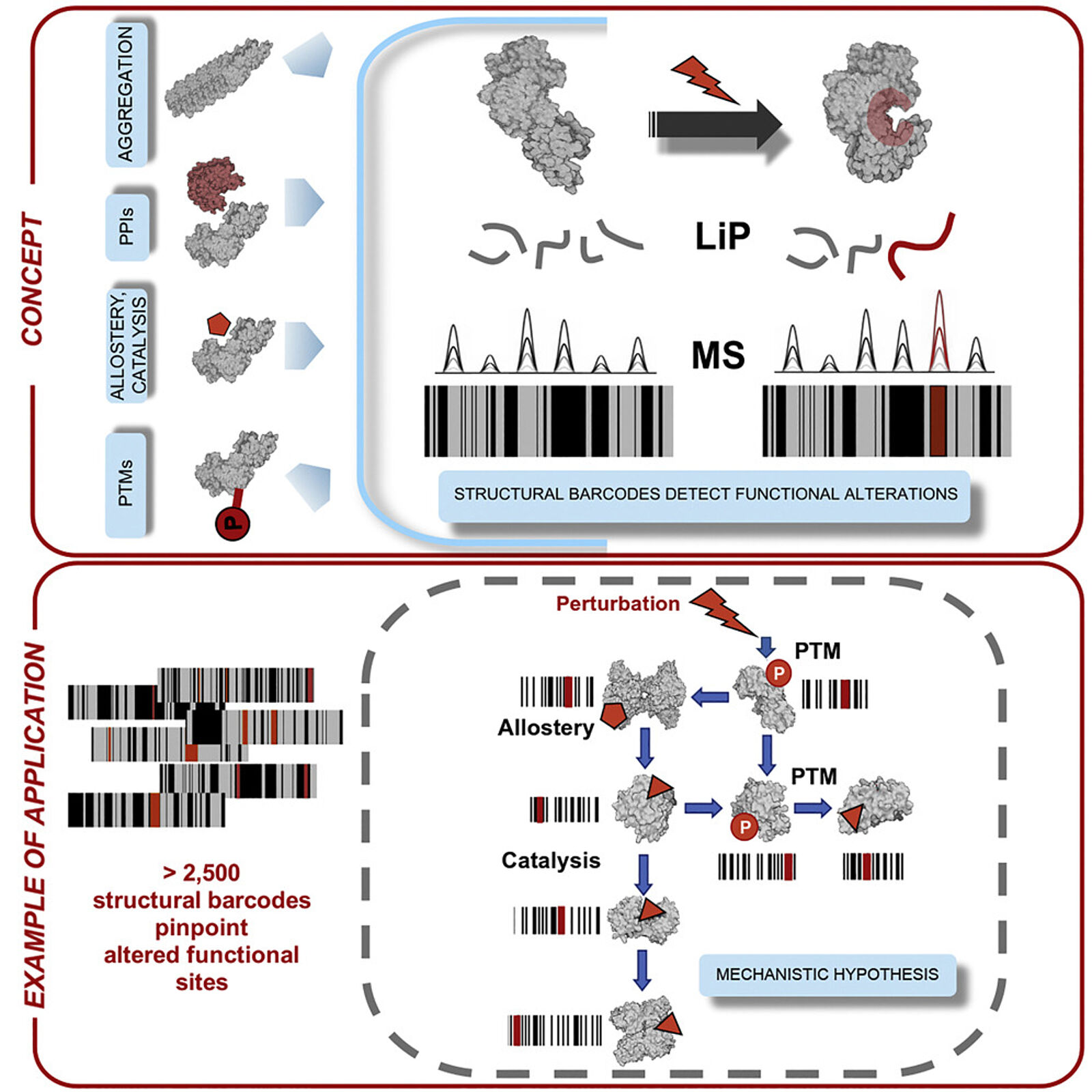
In addition to identifying proteins that have undergone structural changes, the technique is also able to identify what sites within the protein have been altered with a resolution of a few amino acids. This may help to determine how the structural changes are impacting biological processes or to produce mutated forms of the proteins to explore hypotheses about the impact of these structural changes.
References
Cappelletti et. al. Cell (2020), Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ