Background
Whole genome sequencing (WGS) provides comprehensive genomic profiling and may be used to link disease to causal variants [1]. However, the large, all-inclusive data set of genomic sequences can be expensive to produce and is often difficult to store and analyze. This prompts many researchers to instead use targeted next generation sequencing (NGS). Targeted NGS is faster, more cost-effective, and yields a more manageable data set than WGS. Targeting the exome, or protein-coding genes, has revealed many disease-causing variants [2,3].
Both WGS and targeted NGS provide genomic data, which, while valuable, fails to capture changes in gene expression over time or in response to stimuli. RNA sequencing (RNA-seq) measures the transcriptome, a volatile, changing genetic landscape that is influenced by the environment and time. Pairing RNA-seq with WGS can provide researchers with information about how gene variants can lead to changes in gene expression, but performing both techniques is cost-ineffective.
Common methods of NGS, like WGS and targeted NGS, use short-read sequencing, meaning that the resulting sequences (reads) are only 75–300 base pairs in length. Long-read sequencing (LRS) technologies can sequence 10 kb in a single read, using fewer reads to cover the whole genome. LRS provides information regarding long-range interactions and structural variations [4]. Long-read RNA-seq allows researchers to identify isoforms of genes. With short-read sequencing, RNA or protein isoforms can be predicted in areas of dropout, but incorrectly derived isoforms will throw off quantitation. Dainis, et al., used a combination of targeted, long-read, genomic sequencing with targeted, long-read, transcriptomic sequencing to overcome this challenge and provide a comprehensive, cost-effective view of hypertrophic cardiomyopathy (HCM).
HCM is a is a disease in which the myocardium, or heart muscle, hypertrophies, or becomes abnormally thick. This common, complex, genetic disorder is a frequent cause of sudden death in young athletes. Many patients are affected by alterations to either MYBPC3 (cardiac myosin binding protein C) or MYH7 (myosin heavy chain 7). Understanding how MYBPC3 is altered may aid in diagnosis of HCM [5].
The experiment
The researchers focused on a young, female patient with HCM, comparing her sequencing data with 9 additional samples consisting of 6 healthy controls and 3 patients with known HCM-associated point mutations in MYH7. The scientists created genomic and transcriptomic libraries before performing hybridization capture with IDT xGen Lockdown Probes, targeting 10 cardiac genes. Half of the samples were pooled for Pacific Biosciences (PacBio) SMRT sequencing, and the other half were pooled for Oxford Nanopore MinION sequencing. The libraries were pooled and sequenced on 2 flowcells for each sequencing platform, 1 for genomic libraries and 1 for transcriptomic libraries. Reads were aligned to the hg38 reference genome using minimap2 (version 2.10) [6]. Alternative splicing patters were manually identified using the Integrated Genomics Viewer (IGV) [7].
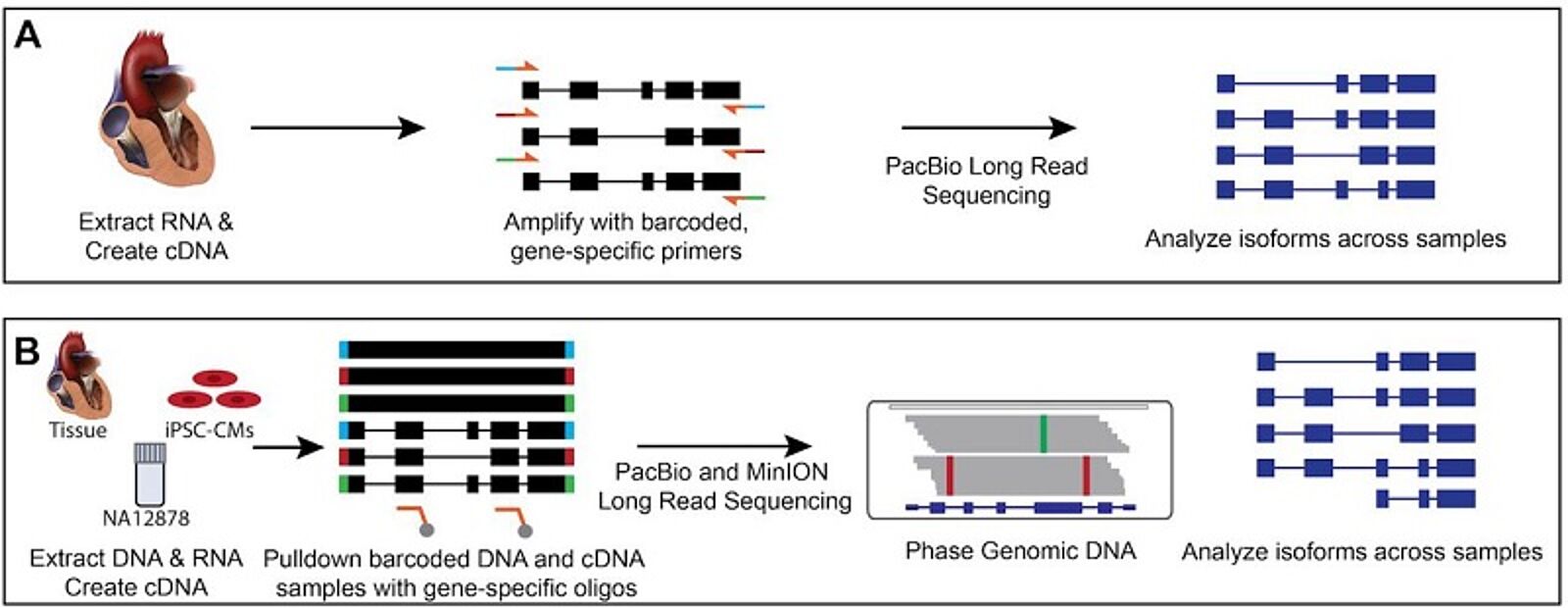
Strategies for investigating alternative splicing and phasing in cardiovascular disease genes
In the first method of phasing cDNA from left ventricular (LV) heart samples (A), Dainis, et al., created cDNA from RNA extracted from frozen LV samples and amplified a transcript of interest (MYBPC3) with barcoded primers specific to the first and last exons. Barcoded samples were pooled and sequenced using Pacific Biosciences SMRT sequencing. This allowed the researchers to phase and compare isoforms across samples. In the second strategy (B), the researchers extracted DNA and RNA from the same samples, which included NA12878, an iPSC-CM cell line, and a left ventricular heart sample. cDNA was created from the RNA, and this cDNA and fragmented genomic DNA was barcoded. IDT xGen oligos specific to the exons of 10 cardiovascular genes of interest were used for target enrichment by hybridization capture. Samples were pooled and sequenced using either PacBio’s SMRT sequencing or Oxford Nanopore Technologies’ MinION sequencing. This allowed the researchers to phase the genomic DNA and cDNA, as well as analyze isoforms across samples.
Results and discussion
The team successfully performed both genomic and transcriptomic sequencing with either PacBio SMRT sequencing or Oxford Nanopore Technologies MinION sequencing technologies. The combination of targeted, long-read, genomic sequencing with targeted, long-read, transcriptomic sequencing identified unexpected alternative protein-isoforms in addition to the predicted alternative isoform resulting from the mutant allele, MYBPC3.
This approach to sequencing allowed researchers to evaluate how genomic variants impact gene and transcript expression and uncover alternative splicing of isoforms. Targeted, long-read sequencing of the genome and transcriptome may provide molecular data that addresses downstream effects of these variants and their contribution to disease, leading the way to new forms of precision medicine.
References
- Bagnall RD, Ingles J, et al. (2018) Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 72(4):419–429.
- Ng SB, Turner EH, et al. (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461(7261):272–276.
- Bamshad MJ, Ng SB, et al. (2011) Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 12(11):745–755.
- Mantere T, Kersten S, et al. (2019) Long-read sequencing emerging in medical genetics. Front Genet. 10:426–426.
- Maron BJ and Maron MS (2013) Hypertrophic cardiomyopathy. Lancet. 381(9862):242–255.
- Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34(18):3094–3100.
- Robinson JT, Thorvaldsdóttir H, et al. (2011) Integrative genomics viewer. Nat Biotechnol. 29(1):24–26.
Copyright 2020 Integrated DNA Technologies. Written by Jessica DeWitt, PhD, Scientific Writer, IDT.